Molecular Simulation of Crystallization
Personnel: M. Hütter, M.J. Ko, N. Waheed, P. Yi
Sponsorship: This work is supported by the Engineering Research Centers Program of the National Science Foundation under NSF Award Number EEC-9731680. (NSF CAEFF) (sponsor contact information)
Macromolecular crystallization is an industrially and scientifically important phenomenon that has defied detailed molecular description to date, despite recent intensified efforts. By focusing separately on the phenomena of nucleation and growth of the crystal phase, we have been able to develop molecular level models that accurately reflect the macroscopically observed phase transformation. Our efforts are two-fold in that they tackle both measuring material properties and structure by microscopic simulations and how such microscopic information is transferred onto continuum-level modeling of complex processes.
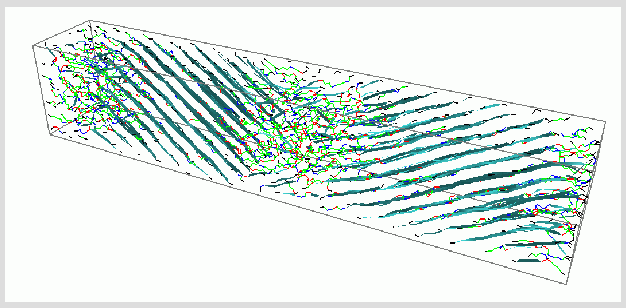
We demonstrated previously that orienting fields are sufficient to accelerate nucleation into the time window accessible by current MD simulations [Polymer 44, 1771 (2003)]. Using the same strategy, we have employed nonequilibrium molecular dynamics to study crystallization of polyethylene from an oriented melt over a range of temperatures [J. Chem. Phys. 121, 2823 (2004)]. The development of crystallinity is monitored simultaneously using molecular based order parameters for density, energy and orientation. We observed a competition between the rate of nucleation, the rate of lamellar thickening, and the rate of chain conformational relaxation in the noncrystalline regions. The temperature dependence of lamellar thickness is in accord with experimental data. At the higher temperature, tilted chain lamellae are observed to form with lamellar interface corresponding approximately to the [201] facet, indicative of the influence of interfacial energy.
In the study of crystal growth, we have implemented molecular dynamics simulations, on alkanes of length C20, C50, and C100, to capture the molecular weight and temperature dependence of crystallization rates for the phase change from a confined melt to a hexagonal close-packed crystal structure [Polymer, in press (2005)]. The growth process for C20 has been decomposed into elementary ordering and melting processes, which suggests the necessity of a secondary nucleation model that incorporates reversible attachment. For C50 and C100, we have been able to quantify growth rates and make qualitative comments about the likelihood of secondary nucleation mechanisms. We have observed the size and shape of a surface nucleus for C50 and C100 systems, which provides insight into the mechanisms involved in alkane crystallization. From the MD data, we have parameterized an analytical crystallization model that predicts crystal growth rates as a function of temperature and molecular weight up to the entanglement molecular weigh, based on relaxation of the chains in the melt and the traditional thermodynamic barrier from classical nucleation theory. For entangled polymers, we have shown that the relevant relaxation time is that of the entangled segment [J Polym. Sci. B. 2005, 43(18), 2468.]; we validate this approach using theory, our simulations, and recent experimental data for polyethylene crystal growth.
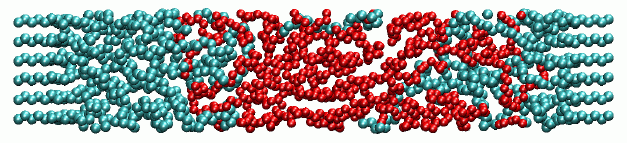
The melt/crystal interface in semi-crystalline polyethylene is studied on an atomistic level using Monte Carlo (MC) simulations. The results show how the melt chains are interwoven with the crystal chains (Fig 2). This is of paramount importance in flow situations, where the interconnection of the two phases is a measure for the transfer of melt deformations to the crystal chains. Measurements of the chain segment orientation reveal that the crystal chains show increasing persistence and orientation closer to the crystal, whereas the melt chains are strongly bent close to the crystal surface. We also determine the interfacial tension of the melt/crystal interface, which is used in continuum-level two-phase models.
Flow-induced crystallization of polymers produces different crystal shapes depending on flow type and strength. To facilitate the development of crystal shape and morphology from molecular level information on nucleation and growth, we developed a continuum-level crystallization model that is capable of distinguishing, for example, row-structures, lamellae, and spherulites. Transient processing conditions can be implemented in a straightforward manner, which is essential to describe shish-kebab growth [Phys. Fluids 17, 014107 (2005)].